

July 2026: Fluorescent Protein Tags
Fluorescent protein fusions are very powerful tools in cell biology. By attaching GFP, mCherry, or related reporters to a protein of interest, researchers can observe where that protein goes, how it moves, and how it responds to perturbations in living cells. When planning experiments using fluorescent proteins, it is important to consider how these tools may introduce artifacts in your experiments and what controls you should include to ensure that any patterns you observe are driven by the protein of interest, not the tag. In both endogenous knock-in and overexpression experiments, fluorescent tags can alter protein folding, trafficking, complex formation, or membrane association, creating mislocalization artifacts that can be mistaken for genuine biology.
This issue appears across experimental systems. In yeast, fluorescently tagged proteins can fail to localize correctly if the fusion disrupts targeting sequences or alters stoichiometry. Even when the tagged protein is expressed from its native locus, the tag itself can interfere with assembly into multiprotein complexes or with retention at membranes and organelles. In bacteria, which have highly constrained cytoplasmic space and specialized secretion or polar localization machinery, bulky fluorescent fusions can be especially problematic. Overexpressed tagged proteins may aggregate, saturate chaperones, or overwhelm native sorting pathways, producing diffuse or ectopic signals (see figure below). In mammalian cells, the problem is equally serious: overexpression can drive nonphysiological accumulation in the ER, Golgi, mitochondria, nucleus, or stress granules, while the fluorescent tag can mask localization signals or create novel interactions that do not occur in the untagged protein.
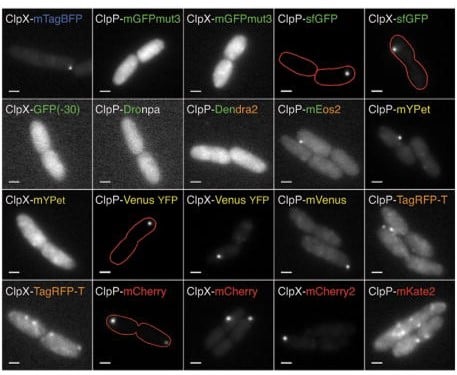
A grid of 20 grayscale fluorescence microscope images showing rod-shaped cells labeled with different fluorescent protein fusions, including several with red outline annotations highlighting cell boundaries or specific structures. The identies of each fusion are listed on the image. Reproduced from: Landgraf D, Okumus B, Chien P, Baker TA, Paulsson J. Segregation of molecules at cell division reveals native protein localization. Nat Methods. 2012 Apr 8;9(5):480-2. doi: 10.1038/nmeth.1955. PMID: 22484850; PMCID: PMC3779060.
A common trap is assuming that visible fluorescence equals correct localization. A fusion may appear to “localize” simply because the protein is overabundant, trapped in a compartment, or misfolded and recognized by quality-control pathways. Conversely, a protein’s true localization may be missed if the tag disrupts targeting or if the fluorescent protein matures slowly relative to the biology being studied. The result can be a misleading image that looks convincing
Experimental Design:
Careful experimental design will help you avoid this trap.
1. Confirm localization with multiple strategies: construct versions of the fusion protein with tags at each terminus and validate localization with orthogonal methods like immunostaining of the native protein or other staining approaches. A particularly strong safeguard is to confirm observed localization using fluorescent proteins from different evolutionary lineages. GFP-like proteins, red fluorescent proteins, and more distantly related reporters can differ in maturation kinetics, oligomerization tendency, brightness, and susceptibility to pH or cellular environment. If a localization pattern is real, it should be reproduced with independent tags that differ in sequence and biophysical behavior.
2. Check whether the tagged protein remains functional. If the fusion cannot complement a deletion or phenocopies a loss-of-function allele, its localization may not be trustworthy.
3. Expression level matters. Endogenous expression is often more reliable than overexpression, because it preserves native stoichiometry and limits artifactual compartment saturation.
SUMMARY:
The broader lesson is simple: fluorescent fusions are tools, not ground truth. They are most informative when paired with controls, independent validation, and healthy skepticism. When working with fluorescent proteins, the image on the screen should be the beginning of the analysis, not the end.