

Research
Current Projects in the Rittenhouse Laboratory
The Rittenhouse lab examines protein-protein, protein-lipid, protein-small molecule, and/or protein-nucleic acid interactions of voltage-gated calcium channels (VGCCs). Disruptions of these interactions lead to certain diseases. This research focus highlights functional aspects of molecular interactions across a spectrum of preparations from established immortalized cell lines to primary neurons, human pancreatic b-cells and human iPSCs. Many of our studies involve measuring activity of molecules in real time using state-of-the art imaging methodologies, secretion assays, and electrophysiological methodologies. Our goals are to reveal how specific molecules interact and function under normal and pathological conditions.
Diseases result from physiological changes in our bodies that adversely affect our health. Ultimately, these disease precipitating changes occur at the molecular level. Every molecule has binding partners that form a functional cassette often used in many cell types. Using this information, we build upon these molecular interactions to predict function at the systems level and identify therapeutic targets for treating brain diseases, such as Alzheimer’s Disease, Stroke, and Schizophrenia.
The Rittenhouse lab is asking three questions that interrogate the regulation of VGCCs in health and disease:
- How do neurotransmitters and second messengers alter VGCC activity?
- Do vicinal palmitoyl groups from palmitoylated proteins bind to VGCCs to change their responsiveness to modulatory mechanisms initiated by neurotransmitters?
- Does the protein Disrupted in Schizophrenia 1 (DISC1) control VGCC levels at synapses?
Project 1: Modulation of voltage-gated Ca2+ channels by neurotransmitters and signaling molecules.
The Rittenhouse lab has a long-standing interest in N-type VGCCs because of their special position in the nervous system. They coordinate electrical activity occurring at the cell membrane with underlying biochemical and transcriptional events. N-VGCCs are found only in nerve cells and neuronally-derived tissues, are associated with the regulation of transmitter synthesis, and release from most presynaptic nerve endings. They are the most extensively modulated VGCCs in the brain in that more pathways exist for their modulation than for any other type.
While plasticity of the brain at the level of complex human behavior is quite obvious, it is also apparent at the cellular and molecular level. One initial site for plasticity occurs with the influx of Ca2+ through VGCCs. Ca2+ influx serves a unique function of interfacing electrical signals with cellular biochemical and transcriptional changes. Variability in VGCC expression levels, in the location of cell-surface expression and in channel activity has profound effects on how much and where Ca2+ enters a nerve cell. This in turn influences the strength of synaptic contacts and on cellular and transcriptional processes. We are currently interrogating how phospholipids and free fatty acid metabolites modulate the N-VGCC using a variety of electrophysiological, imaging, biochemical, and molecular strategies. Superior cervical ganglion neurons (SCG) are used in this study because N-VGCCs are the dominant Ca2+ channel present.
Lab efforts on this project currently focus on two questions:
- How does native N-VGCC activity change when modulated by neurotransmitters, which utilize small lipid signaling molecules such as fatty acids?
- What role does cPLA2a play in neurotransmitter-mediated channel modulation by small lipid signaling molecules?
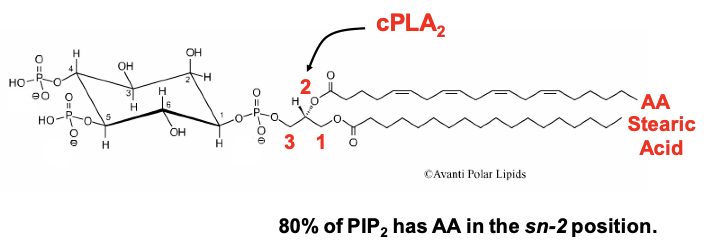
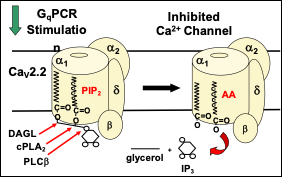
Figure 1: PIP2-AA Model of Phospholipid Metabolism by GqCRs. VGCCs are formed by a protein complex of three subunits: a pore-forming a1-subunit (CaVa1), a transmembrane a2-d subunit, and a cytoplasmic b-subunit (CaVb) that binds to the AID segment of CaVa1’s I-II cytoplasmic linker.
Our findings support the following model: a phospholipid, possibly phosphatidylinositol 4,5-bisphosphate (PIP2) is the source of endogenous polyunsaturated free fatty acid, arachidonic acid (AA), is. AA is normally found as one of the fatty acid tails of PIP2 (Fig. 1). PIP2 binds to N-VGCCs; its presence couples the voltage sensor movement to opening the channel. Muscarinic M1 receptors (M1Rs) inhibit N-VGCC activity by a Gq signaling pathway involving metabolism of the bound phospholipid and thus has served as a model system to examine lipid effects on channels.
The lab currently is interrogating the following hypotheses:
- M1R coupling to Gq activates phospholipase C (PLCb) to remove the inositol head group from the PIP2 associated with each channel. This first step is insufficient to confer inhibition.
- Both diacylglycerol lipase (DAGL) and group IVa phospholipase A2 (cPLA2a) must cleave the two fatty acid tails (normally stearic acid in the sn-1 position and AA in the sn-2 position) from the glycerol backbone to observe VGCC inhibition.
- The two fatty acid tails are predicted to remain bound to the channel. By occupying sites where PIP2’s fatty acid tails normally reside, the free fatty acids block a PIP2 molecule from re-binding to the PIP2 binding site and thus uncouples voltage sensing from channel opening.
This simple model resolves previous conceptual disagreements over the mechanism of N-VGCC inhibition by Gq signaling and provides a framework for pursuing additional questions surrounding Ca2+ channel regulation by lipid molecules.
Additionally, we have interrogated the functional importance of cPLA2a in regulating action potential firing properties. We found that SCG neurons from cPLA2a’s knockout mice exhibit neuronal hyperexcitability, indicating the enzyme provides tonic regulation of phospholipid interaction with multiple types of ion channels.
Significance: Currently, antagonizing cPLA2a is a therapeutic strategy for decreasing excitotoxicity during stroke. The main target of SCG neurons is cerebral blood vessels. These surprising findings raise questions about this therapeutic strategy over the long term since inhibiting cPLA2a could cause severe cerebral vasospasm. Thus, cPLA2a may be a useful target for debilitating transient ischemic attacks.
Project 2: Do vicinal palmitoyl groups from proteins interact with VGCCs to change their responsiveness to modulatory mechanisms initiated by neurotransmitters?
Expression of CaVb2a vs other CaVb subunits appears to block inhibition of N- and L-VGCC activity by M1 muscarinic receptor signaling in sympathetic neurons. Genetic, electrophysiological, biochemical and cell biological methodology are used to:
- Test whether palmitoylation of CaVb2a’s two N-terminus cysteines (C3,C4) are responsible for this block and/or whether additional residues of CaVb2a are involved.
- Test whether the CaVb2a’s palmitoyl groups compete with PIP2 for binding to the channel.
- Identify the lipid interaction domain where the palmitoyl groups interact with VGCCs.
If proven correct, the findings would document a new role for palmitoylation of cytoplasmic proteins – that of serving as a phospholipid mimic that regulates transmembrane proteins.
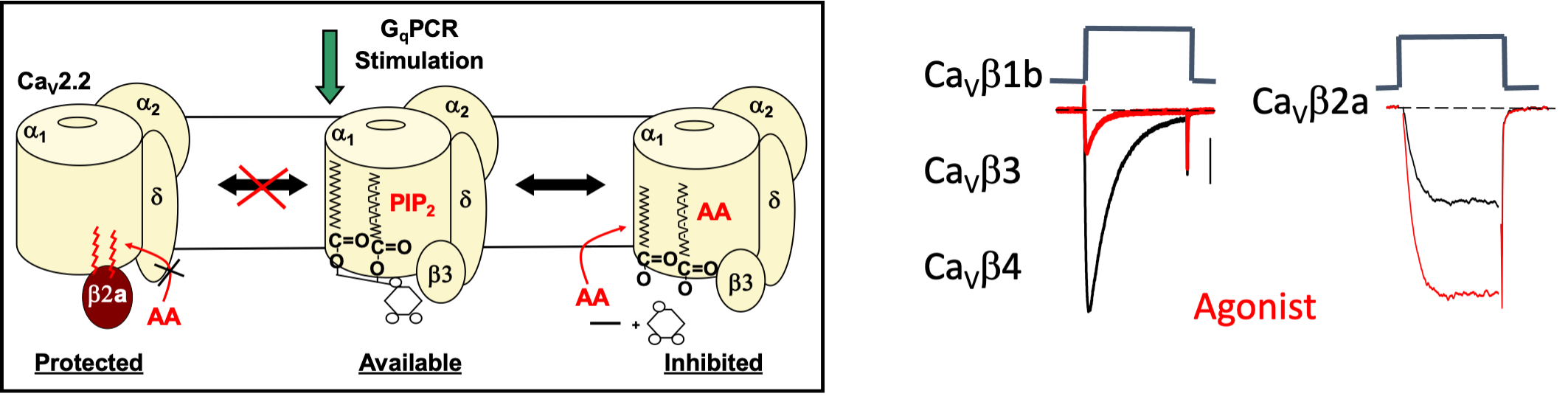
Figure 2. M1R agonist or free AA inhibits N-VGCC activity from recombinant channels made up of a b1, b3, or b4 subunit coexpressed with CaV2.2e, the N-VGCC pore-forming subunit, and a2d1, an accessory subunit. Comparison of N-VGCCs protected with palmitoylated β2a (Protected) to N-VGCCs with β3 that are susceptible (Available) to inhibition by GqPCR signaling (Inhibited). The exaggerated size of PIP2 serves to illustrate the 3 different lipids predicted to interact with N-VGCCs.
In contrast, coexpression with a CaVb2 splice variant, CaVb2a, blocks N-VGCC inhibition revealing a latent enhancement of current. Enhancement occurs at a separate phospholipid -binding site from inhibition. Coexpression of CaV2.2e, and a2d with depalmitoylated CaVb2a restores inhibition, indicating that palmitoylation is necessary for block.
A similar variability in current modulation occurs with a different class of channels, the L-VGCCs CaV1.3, when co-expressed with different CaVb subunits, indicating that CaVb2a interacts with other pore-forming subunits to block VGCC inhibition by M1Rs (Fig. 3).
Figure 3. L-VGCCs CaV1.3, when co-expressed with different CaVb subunits, indicating that CaVb2a interacts with other pore-forming subunits to block VGCC inhibition by M1Rs.
Significance: These studies reveal a new role for protein palmitoylation; that of regulating VGCC modulation. Moreover, our findings raise the possibility that the palmitoyl residues of CaVb2a directly interact with N-VGCCs at the phospholipid binding site. If the palmitoyl groups substitute for PIP2’s fatty acid tails, no phospholipid metabolism would occur upon M1R signaling because no phospholipid is bound: the channels are protected from inhibition. Palmitoylation-depalmitoylation events allow M1R responses to toggle between current inhibition and enhancement from moment to moment depending on the activity of phospholipases, acyl hydrolases, and palmitoyl acyl transferases. Moreover, sequentially palmitoyaled proteins serving as a phospholipid mimic provides a unique mechanism for regulating transmembrane proteins.
Project-3: Do vicinal palmitoyl groups of other proteins bind to VGCCs?
As proof of principle, other proteins that are sequentially palmitoylated are predicted to minimize VGCC inhibition by GqPCRs, similar to CaVb2a, by interacting with their phospholipid binding sites. To test this possibility, we interrogated L-VGCCs that couple to the secretion machinery of pancreatic b-cells. These cells were chosen for their translational relevance. A high circulating palmitate level in blood leads to decreased insulin secretion and increased susceptibility to developing Type 2 Diabetes (T2D). Disruption of interactions between protein palmitoyl groups and VGCCs results in decreased insulin release. These changes may underlie increased susceptibility to T2D.
Project-4: The Role of VGCC regulation by DISC1 in Schizophrenia
Schizophrenia is a complex, devastating mental illness that affects 1% of the population worldwide. Its onset occurs as the result of genetic predisposition, environmental factors and their interaction. Three major systemic pathologies are observed in schizophrenic patientss including, cognitive dysfunction, altered release of cytokines from immune cells, metabolic syndrome expressed by Type 2 Diabetes (T2D) and insulin insensitivity. Surprisingly, all three systems exhibit altered secretion, raising the possibility that schizophrenia is a disease of secretion.
A connection between T2D and psychiatric disorders has been recognized for more than a century: in 1879, psychiatrist Henry Maudsley, MD, noted that “diabetes is a disease which often shows itself in families in which insanity prevails”. Although the co-occurrence of psychiatric disorders and T2D has been well documented over the years, the cause of this association remains unknown. As antipsychotic medications came into use in the 20th century, it became apparent that many antipsychotic medications themselves also increase the risk of diabetes. This increased risk often associates with the weight gain that frequently occurs with antipsychotic use, but carefully controlled studies also reveal an increased risk for T2D independent of obesity or other lifestyle factors, suggesting involvement of a genetic component(s). Understanding molecular mechanisms common to both diseases may reveal a parsimonious pathway that contributes to both psychosis and diabetes. Thus, identifying common molecular markers poses a major medical challenge, but a critical one, for developing an understanding of disease mechanism as the morbidity and mortality associated with T2D and psychiatric disorders are a major health concern worldwide.
The L-VGCC pore-forming subunit CACNA1C (CaV1.2), its accessory subunit CACNB2 (CaVb2), and mir137, the microRNA that controls their expression, repeatedly score highest as schizophrenia susceptibility genes by GWAS. These same two subunits were found in a GWAS for T2D. Notably, central neurons, T-cells, and b-cells all express CaV1.2 and CaVb2. Moreover, the CaVb2 splice variant is lipid modified as in SCG neurons: two palmitoyl groups are bound to sequential (vicinal) cysteine residues in the N-terminus of the splice variant CaVb2a.We are particularly interested in monitoring their expression and activity because L-VGCCs initiate secretion from each cell type. We are also studying the gene, Disrupted in Schizophrenia 1 (DISC1), because loss of this protein increases VGCC instability. DISC1 was identified as a schizophrenia susceptibility gene in a family with high penetrance for major psychiatric disorders. In collaboration with the Diabetes Center of Excellence, we found decreased glucose stimulated insulin secretion, intracellular Ca2+ signaling and L-VGCC activity due to a dominant-negative effect of truncated human DISC1 expression in mouse and human donor pancreatic b-cells.
Significance: DISC1 has independent but similar actions in neurons and b-cells: that of regulating the levels of VGCCs, which couple excitation to secretion. Moreover, these findings suggest a disturbing functional linkage between major psychotic disorders (bipolar disorder, major depression, and schizophrenia) and T2D. Indeed, diabetic individuals have higher rates of major depression, suggesting underlying molecular links between brain and pancreatic islets. If a subset of patients with major psychosis have increased susceptibility to T2D due to disruption of the same cellular pathway in neurons and b-cells, then the reverse may also be true. If so, a subpopulation of the millions of people worldwide with T2D will be at risk for major psychosis, an alarming possibility that warrants further investigation. We are particularly interested in screening for changes in VGCC expression and activity in immune cells, hiPSC b-cells, and donor b-cells from schizophrenic patients and in mouse models of schizophrenia.
Currently, we are testing for changes in DISC1, VGCC subunits, and immune markers to answer a series of questions:
- Does mutant DISC1 result in a down regulation of VGCC subunits in pancreatic b-cells?
- Is secretion machinery disrupted by mutant DISC1 in pancreatic b-cells?
- Are immune cell DISC1 levels altered in idiopathic schizophrenic patients?