

Research
Early studies on phage recombination functions
Early in my career, I studied components of recombination systems from bacteriophage lambda and phage P22. These studies included an early description of the single-stranded DNA annealing (SSDA) function of phage P22 called Erf (for essential recombination function). It is a functional analog of the well-known λ Beta annealase of the λ Red system, and promotes annealing of two complementary single-stranded DNAs in vitro. The λ Beta protein can substitute for Erf during a P22 phage infection in Salmonella typhimurium, and has a ring like structure with C-terminal protrusions as seen under an electron microscope (like λ Beta). Its domain structure has been analyzed both biochemically and genetically (1).
In addition, other recombination-promoting phage functions were an interest of mine at that time, including phage-encoded modifiers of the host RecBCD enzyme (aka ExoV). While RecBCD is a major component of the main route of homologous recombination in E. coli and S. typhimurium, it is also a powerful ATP-dependent exonuclease. Recombination via RecBCD is dependent on the enzyme’s encounter with the hotspot initiator Chi sequences (GCTGGTGG), which alters the enzymatic properties of RecBCD such that the enzyme generates 3’ ssDNA recombinogenic tails at dsDNA ends. However, both phage λ and P22 do not have Chi sites and thus cannot take advantage of the recombinogenic potential of RecBCD, and instead, are subject to the destructive action of RecBCD’s dsDNA exonuclease. To counteract the action of RecBCD, both phage λ and P22 encode anti-RecBCD modifiers, which both protect their chromosomes for exonucleolytic attack and allow their recombination systems to act unimpeded. However, each anti-RecBCD function is different. While the λ Gam protein binds to RecBCD and prevents the enzyme from binding dsDNA ends (2, 3), the anti-RecBCD function from phage P22 (called Abc) binds to the RecC subunit, inhibits Chi activity, and hijacks the exonuclease activity of RecBCD to promote phage recombination. Phage P22 does not have an λ-like exonuclease; instead, it uses a modified RecBCD exonuclease function to promote phage recombination (4-6). The Gam protein plays an important role in recombineering by protecting linear dsDNA substrates following electroporation into E. coli, giving time and opportunity for the λ Red system to work efficiently.
Discovery of gene replacement in E. coli using the λ phage Red recombination system.
E. coli gene replacement technologies in the 1990s consisted of two major avenues. One was to transform a non-replicating plasmid that contained a mutant version of the target gene. This was often a very time-consuming process that relied on endogenous recombination systems, occurred at low frequencies, and required a second counter-selection step to resolve the co-integrant. The second method was the use of recombination–proficient strains of E. coli (sbcA, recBC sbcB15, or recD) that increased the frequency of transformation with linear dsDNA species, albeit to a low and variably efficient rate. It became evident at the time that the genotypes of these recombination–proficient strains were similar: they all resulted in inactivation of the host RecBCD dsDNA exonuclease, and induced an alternate homologous recombination pathway. These were both properties of the phage lambda Red pathway that I had been studying at the time. Thus, I tested whether λ Red and Gam could be used to promote recombination of linear dsDNAs with the bacterial chromosome, by producing these functions from a plasmid or by substituting the chromosomal recBCD region with the Plac-red-gam functions. To my surprise, linear dsDNA transformation frequencies were 100 times higher with λ Red and Gam expression relative to recD and recBC sbcB15 strains being used for gene replacement at the time (7). This was the first demonstration of the λ Red gene replacement methodology. I shared my results with both the Court and Wanner labs, who later showed that such recombination frequencies only requires ~ 50 bp of flanking homologies, a result I had discovered as well, by expressing the phage functions from a stronger promoter (Ptac, instead of Plac). The use of such short homologies had already been demonstrated in the Stewart lab, using the RecET recombination system from the rac prophage. The process became known as “recombineering” and has revolutionized the methods of bacterial genetic engineering. Some of my contributions to this field are highlighted in primary articles and reviews listed below (8-13).
Genetic engineering of pathogenic bacteria
In 2003, I was able to demonstrate the successful use of the Red system in enterohemorrhagic E. coli (EHEC), as well as enteropathogenic E. coli (EPEC) (13). The λ Red-producing plasmid for performing recombineering in these pathogens, pKM208, has been cited in numerous articles and is available form the addgene.com plasmid repository website. Other work representing recombineering of EHEC strains from my lab are listed below (14-17). In 2007, my research took a turn toward the development of genetic engineering strategies for Mycobacterium tuberculosis, highlighted by an extensive and continuing collaboration with colleague Christopher Sassetti here in the Microbiology department. Studies toward this end and current work regarding DNA mismatch repair mechanisms in mycobacteria can be found below.
1) Murphy KC, Casey L, Yannoutsos N, Poteete AR, Hendrix RW. Localization of a DNA-binding determinant in the bacteriophage P22 Erf protein. J Mol Biol. 1987 Mar 5;194(1):105-17. doi: 10.1016/0022-2836(87)90719-4. PubMed PMID: 3612797.
2) Murphy KC. Lambda Gam protein inhibits the helicase and chi-stimulated recombination activities of Escherichia coli RecBCD enzyme. J Bacteriol. 1991 Sep;173(18):5808-21. doi: 10.1128/jb.173.18.5808-5821.1991. PubMed PMID: 1653221; PubMed Central PMCID: PMC208314.
3) Murphy KC. The lambda Gam protein inhibits RecBCD binding to dsDNA ends. J Mol Biol. 2007 Aug 3;371(1):19-24. doi: 10.1016/j.jmb.2007.05.085. Epub 2007 Jun 2. PubMed PMID: 17583735.
4) Murphy KC. Biochemical characterization of P22 phage-modified Escherichia coli RecBCD enzyme. J Biol Chem. 1994 Sep 9;269(36):22507-16. PubMed PMID: 8077199. select
5) Murphy KC, Lewis LJ. Properties of Escherichia coli expressing bacteriophage P22 Abc (anti-RecBCD) proteins, including inhibition of Chi activity. J Bacteriol. 1993 Mar;175(6):1756-66. doi: 10.1128/jb.175.6.1756-1766.1993. PubMed PMID: 8383665; PubMed Central PMCID: PMC203970.
6) Murphy KC. Bacteriophage P22 Abc2 protein binds to RecC increases the 5' strand nicking activity of RecBCD and together with lambda bet, promotes Chi-independent recombination. J Mol Biol. 2000 Feb 18;296(2):385-401. doi: 10.1006/jmbi.1999.3486. PubMed PMID: 10669596.
7) Murphy KC. Use of bacteriophage lambda recombination functions to promote gene replacement in Escherichia coli. J Bacteriol. 1998 Apr;180(8):2063-71. doi: 10.1128/JB.180.8.2063-2071.1998. PubMed PMID: 9555887; PubMed Central PMCID: PMC107131.
8) Murphy KC, Campellone KG, Poteete AR. PCR-mediated gene replacement in Escherichia coli. Gene. 2000 Apr 4;246(1-2):321-30. doi: 10.1016/s0378-1119(00)00071-8. PubMed PMID: 10767554.
9) Murphy KC, Marinus MG. RecA-independent single-stranded DNA oligonucleotide-mediated mutagenesis. F1000 Biol Rep. 2010 Jul 22;2:56. doi: 10.3410/B2-56. PubMed PMID: 20711416; PubMed Central PMCID: PMC2920528.
10) Murphy KC. Targeted chromosomal gene knockout using PCR fragments. Methods Mol Biol. 2011;765:27-42. doi: 10.1007/978-1-61779-197-0_2. PubMed PMID: 21815084.
11) Murphy KC. Phage recombinases and their applications. Adv Virus Res. 2012;83:367-414. doi: 10.1016/B978-0-12-394438-2.00008-6. Review. PubMed PMID: 22748814.
12) Murphy KC. λ Recombination and Recombineering. EcoSal Plus. 2016 May;7(1). doi: 10.1128/ecosalplus.ESP-0011-2015. Review. PubMed PMID: 27223821.
13) Murphy KC, Campellone KG. Lambda Red-mediated recombinogenic engineering of enterohemorrhagic and enteropathogenic E. coli. BMC Mol Biol. 2003 Dec 13;4:11. doi: 10.1186/1471-2199-4-11. PubMed PMID: 14672541; PubMed Central PMCID: PMC317293.
14) Savage PJ, Leong JM, Murphy KC. Rapid allelic exchange in enterohemorrhagic Escherichia coli (EHEC) and other E. coli using lambda red recombination. Curr Protoc Microbiol. 2006 Jan;Chapter 5:Unit5A.2. doi: 10.1002/9780471729259.mc05a02s00. PubMed PMID: 18770591.
15)Campellone KG, Roe AJ, Løbner-Olesen A, Murphy KC, Magoun L, Brady MJ, Donohue-Rolfe A, Tzipori S, Gally DL, Leong JM, Marinus MG. Increased adherence and actin pedestal formation by dam-deficient enterohaemorrhagic Escherichia coli O157:H7. Mol Microbiol. 2007 Mar;63(5):1468-81. doi: 10.1111/j.1365-2958.2007.05602.x. PubMed PMID: 17302821; NIHMSID:NIHMS228375.
16) Murphy KC, Ritchie JM, Waldor MK, Løbner-Olesen A, Marinus MG. Dam methyltransferase is required for stable lysogeny of the Shiga toxin (Stx2)-encoding bacteriophage 933W of enterohemorrhagic Escherichia coli O157:H7. J Bacteriol. 2008 Jan;190(1):438-41. doi: 10.1128/JB.01373-
17) Carone BR, Xu T, Murphy KC, Marinus MG. High incidence of multiple antibiotic resistant cells in cultures of in enterohemorrhagic Escherichia coli O157:H7. Mutat Res. 2014 Jan;759:1-8. doi: 10.1016/j.mrfmmm.2013.11.008. Epub 2013 Dec 18. PubMed PMID: 24361397; PubMed Central PMCID: PMC3913999.
Genetic engineering of M. tuberculosis using ORBIT
Oligo-mediated Recombineering followed by Bxb1 phage Integrase targeting We have recently developed a new system for making gene knockouts, fusions, and promoter replacements in Mycobacteria. The methodology is called ORBIT, for Oligo-mediated Recombineering followed by Bxb1 phage Integrase Targeting (1). The system was built based on the extensive work of Graham Hatfull and his students on both site-specific recombination system of the Bxb1 phage and the RecET recombineering systems of the Che9c phage (2-10). In the ORBIT protocol, both the RecT annealase and the Bxb1 Integrase proteins are expressed from a plasmid in either M. smegmatis or M. tuberculosis. The cells are then electroporated with an oligo that contains the Bxb1 phage attP site (GGTTTGTCTGGTCAACCACCGCGGTCTCAGTGGTGTACGGTACAAACC), flanked on both sides with 70 base pairs of target DNA. Along with this oligo, the cells are co-electroporated with a non-replicating plasmid (oriM-) that contains the Bxb1 attB site and a drug-resistance marker. During the outgrowth period, RecT promotes annealing of the oligo to the replication fork, where after additional replication of the region, the attP site is generated in the chromosome, its position dictated by the selection of the flanking sequences in the oligo. In the same outgrowth period, the Bxb1 integrase promotes site-specific integration of the plasmid into the attP site, allowing for selection of the event by virtue of the drugR marker on the plasmid. In effect, an oligo and a pre-existing plasmid are transformed into Mtb, and the knockout is found after the outgrowth is plated on antibiotic-selection plates. No cloning of Mtb sequences is required for generation of the allele-exchange substrate, and the frequencies of gene replacement are often between 50-100%. For knockouts, the oligo is designed so that the attP site replaces the target gene; for C-terminal fusions, the oligo is designed so that the attP site is placed just before the stop codon. The type of fusion desired is dependent on which of the numerous attB-containing integration plasmids is chosen(e.g., eGFP, Flag, SNAP, etc). The RecT-Integrase expressing plasmid and all the ORBIT integration plasmids are available from the plasmid repository website addgene.com. For updates, helpful hints on designing the oligos, and trouble shooting for the ORBIT protocol, see Resources page.
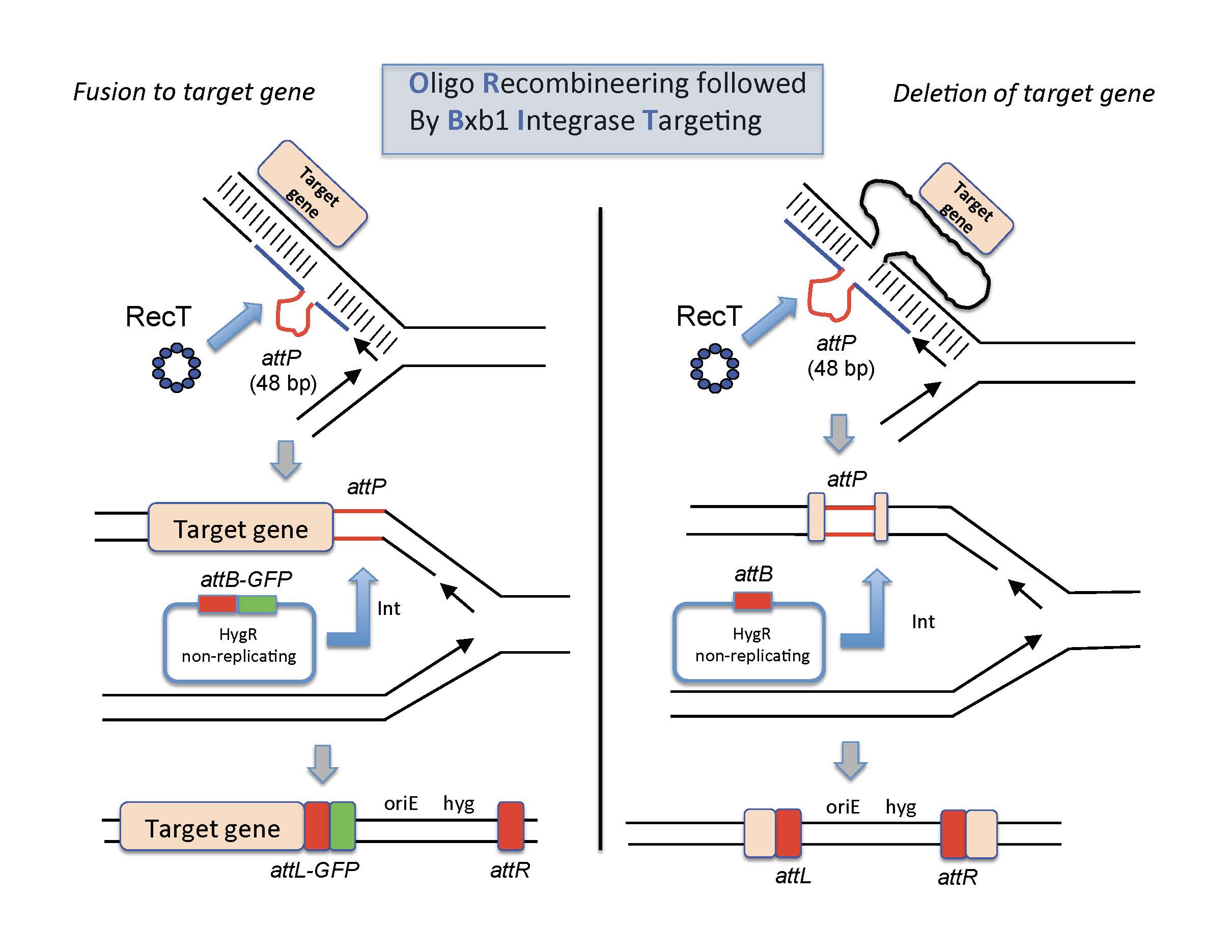
ORBIT-promoted gene alteration. The ORBIT process is initiated at the replication fork. An oligomer containing a single-stranded version of the Bxb1attP site (top pictures, red lines) is coelectroporated with an attB-containing nonreplicating plasmid into a mycobacterial host cell expressing both RecT annealase and Bxb1 Integrase. RecT promotes annealing of the oligonucleotide to the lagging strand template. Following DNA replication through this region, an attP site is formed in the chromosome (middle pictures). In the same outgrowth period, Bxb1 Integrase promotes site-specific insertion of the plasmid into the chromosome (attB x attP). (Left side) The oligonucleotide is designed such that attP is inserted just before the stop codon. The integration event fuses the GFP tag in-frame to the 3’end of the target gene (with an attL site in frame between them); the recombinant is selected for by HygR. (Right side) The oligonucleotide is designed such that attP replaces the target gene and the plasmid integration event allows hygromycin resistance to be used to select for the knockout.
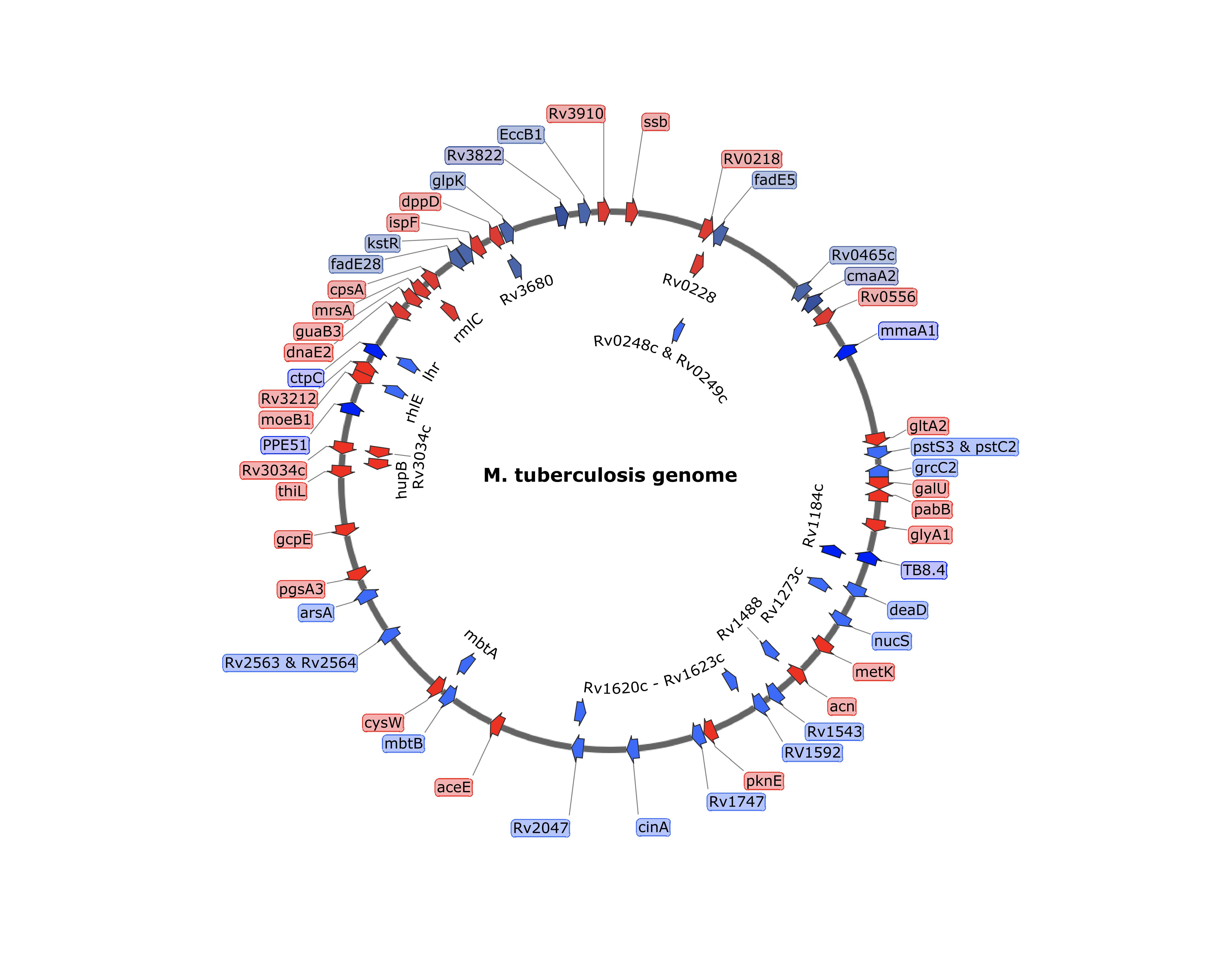
ORBIT-generated insertions and deletions in M. tuberculosis. Target gene deletions and tags were placed at a variety of positions in the chromosomes of M. tuberculosis. In most cases, the oligos contained an attP site flanked by 70 bases of target homology. Insertions (either DAS+4, GFP or His-Flag) are shown in Red and deletions are shown in blue.
References
1. Murphy KC, Nelson SJ, Nambi S, Papavinasasundaram K, Baer CE, Sassetti CM. 2018. ORBIT: a new paradigm for genetic engineering of mycobacterial chromosomes. mBio 9:e01467-18. https://doi.org/10.1128/mBio .01467-18.
2. Kim AI, Ghosh P, Aaron MA, Bibb LA, Jain S, Hatfull GF. 2003. Mycobacteriophage Bxb1 integrates into the Mycobacterium smegmatis groEL1gene. Mol Microbiol 50:463– 473. https://doi.org/10.1046/j.1365-2958.2003.03723.x.
3. Mediavilla J, Jain S, Kriakov J, Ford ME, Duda RL, Jacobs WR, Jr, Hendrix RW, Hatfull GF. 2000. Genome organization and characterization of mycobacteriophage Bxb1. Mol Microbiol 38:955–970. 4. Ghosh P, Kim AI, Hatfull GF. 2003. The orientation of mycobacteriophage Bxb1 integration is solely dependent on the central dinucleotide of attP and attB. Mol Cell 12:1101–1111. https://doi.org/10.1016/S1097-2765 (03)00444-1.
5. Ghosh P, Pannunzio NR, Hatfull GF. 2005. Synapsis in phage Bxb1 integration: selection mechanism for the correct pair of recombination sites. J Mol Biol 349:331–348. https://doi.org/10.1016/j.jmb.2005.03.043.
6. Ghosh P, Wasil LR, Hatfull GF. 2006. Control of phage Bxb1 excision by a novel recombination directionality factor. PLoS Biol 4:e186. https://doi .org/10.1371/journal.pbio.0040186.
7. Savinov A, Pan J, Ghosh P, Hatfull GF. 2012. The Bxb1 gp47 recombination directionality factor is required not only for prophage excision, but also for phage DNA replication. Gene 495:42–48. https://doi.org/10 .1016/j.gene.2011.12.003.
8. Singh S, Ghosh P, Hatfull GF. 2013. Attachment site selection and identity in Bxb1 serine integrase-mediated site-specific recombination. PLoS Genet 9:e1003490. https://doi.org/10.1371/journal.pgen.1003490.
9. van Kessel JC, Hatfull GF. 2007. Recombineering in Mycobacterium tu- berculosis. Nat Methods 4:147–152. https://doi.org/10.1038/nmeth996. 10. van Kessel JC, Hatfull GF. 2008. Efficient point mutagenesis in mycobac- teria using single-stranded DNA recombineering: characterization of antimycobacterial drug targets. Mol Microbiol 67:1094 –1107. https://doi .org/10.1111/j.1365-2958.2008.06109.x.
Mycobacterial DNA Mismatch Repair
The MutSL mismatch repair (MMR) systems in bacteria and eukaryotes correct base pair mismatches made at the replication fork to maintain genome stability . Despite lacking the mutSL genes, most actinobacterial species, including Mycobacterium tuberculosis, exhibit spontaneous mutation rates similar to MutSL-bearing bacteria, suggesting the existence of an alternative MMR system. It has been shown that the EndoMS/NucS protein from actinobacterium Corynebacterium glutamicum is a mismatch-specific endonuclease that binds to and corrects G-T, G-G, and T-T mismatches in vitro (1,2). In addition, previous reports have shown that deletion of the nucS gene in M. smegmatis results in a strain with a mutator phenotype, suggesting it is part of a newly discovered mycobacterial MMR system (3). We have been performing genetic analyses of both M. smegmatis and M. tuberculosis strains deleted of nucS to explore the mechanism of MMR. We use recombineering techniques to deliver specific types of mismatched bases to the replicating chromosome, and then follow the fate of chromosomal repair using correction of a defective hygromycin-resistance gene (Flores & Murphy, manuscript in preparation). By using this system, and other types of genetic analyses, along with biochemical analysis of the purified NucS protein, we hope to uncover the mechanism of this newly discovered MMR system.
References:
1) Ishino S, Skouloubris S, Kudo H et al. (2018) Activation of the mismatch-specific endonuclease EndoMS/NucS by the replication clamp is required for high fidelity DNA replication. Nucleic acids research 46 (12):6206-6217. doi:10.1093/nar/gky460
2) Takemoto N, Numata I, Su'etsugu M et al. (2018) Bacterial EndoMS/NucS acts as a clamp-mediated mismatch endonuclease to prevent asymmetric accumulation of replication errors. Nucleic acids research 46 (12):6152-6165. doi:10.1093/nar/gky481 3) Castañeda-García, A., Prieto, A., Rodríguez-Beltrán, J. et al. (2017) A non-canonical mismatch repair pathway in prokaryotes. Nat Commun 8, 14246, https://doi.org/10.1038/ncomms14246
Single nucleotide polymorphisms (SNPs) transfer in mycobacteria
Mycobacterial Recombineering techniques, and the recently described ORBIT protocol, allow for the generation of gene knockouts and fusions in both M. smegmatis and M. tuberculosis. Less often employed in mycobacterial recombineering is the use of oligonucleotides, which requires only the action of the RecT annealase to align oligos to ssDNA regions of the replication fork, for subsequent incorporation into the chromosome. Despite the higher frequency of such events relative to dsDNA-promoted recombineering, oligo-mediated changes generally suffer from the disadvantage of not being selectable, thus making them harder to isolate. Oligo-mediated recombineering is most suited for the verification of SNPs suspected of being associated with spontaneous drug-resistance, as was done by us in collaboration with Ioerger an Sacchettini (1), or for the transfer of SNPs from clinical isolates to laboratory strains of mycobacteria, to test their relevance to a specific disease phenotype. In the later case, finding the SNP-containing isolate is problematic because of the lack of selectio in most cases of SNP transfer. Work in my lab continues to optimize the methodology for SNP transfer in M. tuberculosis using RecT-promoted oligo-mediated recombineering. First, we find that co-electroporation with an oligo the repairs a defective hygromycin-resistant gene at the L5 site can be used to screen colonies that are competent for DNA pickup, a scheme first described by Van Kessel and Hatfull (2). Screening the HygR candidates for the SNP-of-interest by PCR-product sequencing greatly improves the chances of finding successful SNP transfers. Secondly, use of oligos that avoid the creation of mismatches G-T, G-G, and T-T, following annealing of the oligo to the lagging strand template, or by adding additional mismatches in the vicinity of such mismatches, greatly improves the rate of SNP transfer. We are continuing to optimize the system by creating SNPs in known biosynthetic genes that can be targets for use as the “selectable” oligo.
1) Ioerger TR, O'Malley T, Liao R, Guinn KM, Hickey MJ, Mohaideen N, Murphy KC, Boshoff HI, Mizrahi V, Rubin EJ, Sassetti CM, Barry CE 3rd, Sherman DR, Parish T, Sacchettini JC. Identification of new drug targets and resistance mechanisms in Mycobacterium tuberculosis. PLoS One. 2013;8(9):e75245. doi: 10.1371/journal.pone.0075245. eCollection 2013. PubMed PMID: 24086479; PubMed Central PMCID: PMC3781026.
2) van Kessel JC, Hatfull GF. Efficient point mutagenesis in mycobacteria using single-stranded DNA recombineering: characterization of antimycobacterial drug targets. Mol Microbiol. 2008 Mar;67(5):1094-107. doi: 10.1111/j.1365-2958.2008.06109.x. Epub 2008 Jan 22. PMID: 18221264.
M. tuberculosis Hypomorphic Library Construction
Tuberculosis remains one of the deadliest scourges of all time, killing 1.4 million people in 2019. While a drug regimen exists to treat infected individuals, compliance in high burden countries remains a problem, and multiple- and extensively-drug resistant strains of Mycobacterium tuberculosis (MTb) are appearing at alarming rates. We need new drugs to work at an accelerated rate and to combat the onslaught of MTb drug-resistant strains. In collaboration with Chris Sassetti in our department, our group at UMass has been instrumental in a Gates-funded project that involves collaborators at the Harvard School of Public Health (Rubin lab), Weill Cornell Medical College (Schnappinger and Ehrt labs), and the Broad Institute in Cambridge, MA (Hung lab) to construct a hypomorphic library of MTb strains, where every essential function is tagged at the C-terminal end with a degradation tag. Controlled proteolysis allows lowering the amount of the essential function in vivo, where the bacterium still grows but becomes susceptible to chemical agents in 96 well plates. This target-based whole cell screen (TB-WCS) approach allows one to increase success of finding a compound-target hit, which serves as a potential leading candidates for drug development. In addition, this scheme comes with the added feature that the mechanism of action of an active compound has a high probability of being linked to a particular pathway or gene. The hypomorphic library was constructed by 1) expressing the target gene as a C-terminal tagged Flag-DAS-4 fusion form the L5 phage integration site; 2) knocking out the endogenous target gene using mycobacterial recombineering and 3) expressing various levels of an SspB protein from an anhydrotetracycline-dependent promoters. The SspB protein binds to the DAS+4 tag and delivers the targeted protein to the proteasome. The use of this library to find new classes of M. tuberculosis drugs has been described by Johnson et al (see below).
Johnson EO, LaVerriere E, Office E, Stanley M, Meyer E, Kawate T, Gomez JE, Audette RE, Bandyopadhyay N, Betancourt N, Delano K, Da Silva I, Davis J, Gallo C, Gardner M, Golas AJ, Guinn KM, Kennedy S, Korn R, McConnell JA, Moss CE, Murphy KC, Nietupski RM, Papavinasasundaram KG, Pinkham JT, Pino PA, Proulx MK, Ruecker N, Song N, Thompson M, Trujillo C, Wakabayashi S, Wallach JB, Watson C, Ioerger TR, Lander ES, Hubbard BK, Serrano-Wu MH, Ehrt S, Fitzgerald M, Rubin EJ, Sassetti CM, Schnappinger D, Hung DT. Large-scale chemical-genetics yields new M. tuberculosis inhibitor classes. Nature. 2019 Jul;571(7763):72-78. doi: 10.1038/s41586-019-1315-z. Epub 2019 Jun 19. PMID: 31217586.