


Fatty Oxidation Disorders
Genetic disorders of mitochondrial fatty acid oxidation (FAO) represent a relatively common class of metabolic disorders, exceeding more than 1 in 15,000 newborns. These disorders are associated with genes involved in the mitochondrial b-oxidation of fatty acids, which provide fuel once glycogen stores are depleted during prolonged fasting or during times of increased energy demands and physiological stress.
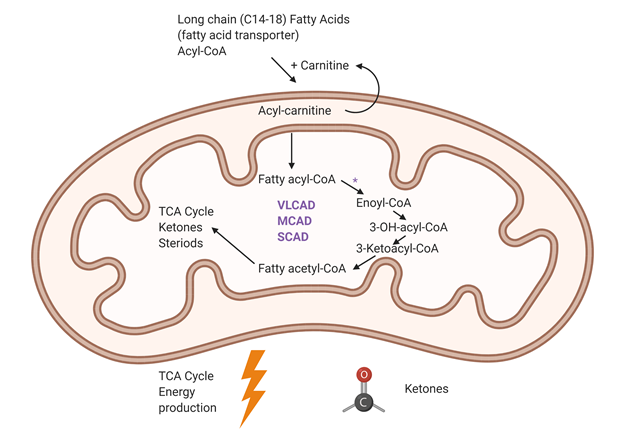
Importantly, the first step of Fatty acid oxidation is catalyzed by an acyl-CoA dehydrogenase. This rate limiting function is performed by a family of enzymes that differ in their substrate specificity based on the carbon chain length of the acyl CoA molecule. The acyl-CoA dehydrogenases (ACDs) are a family of 5 mitochondrial enzymes involved in fatty acid and amino acid metabolism that catalyze the transfer of electrons from various acyl-CoA esters to electron transfer flavoprotein. Very long, medium and short chain acyl-CoA dehydrogenases (VLCAD, MCAD and SCAD) catalyze the first step in the b-oxidation cycle with substrate specificities of 16-, 8- and 4- carbon chains, respectively.
Currently our lab is applying rAAV-based transduction of liver and skeletal muscle for those FAO deficiencies that are most common in humans: medium chain acyl CoA dehydrogenase (MCAD) and very long chain acyl CoA dehydrogenase (VLCAD). Specifically, the feasibility of molecular and biochemical correction of VLCAD and MCAD deficiencies is tested in mouse models of these deficiencies.