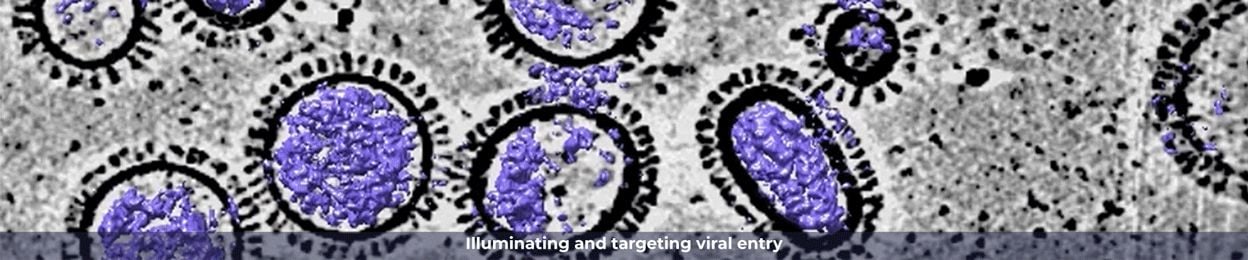


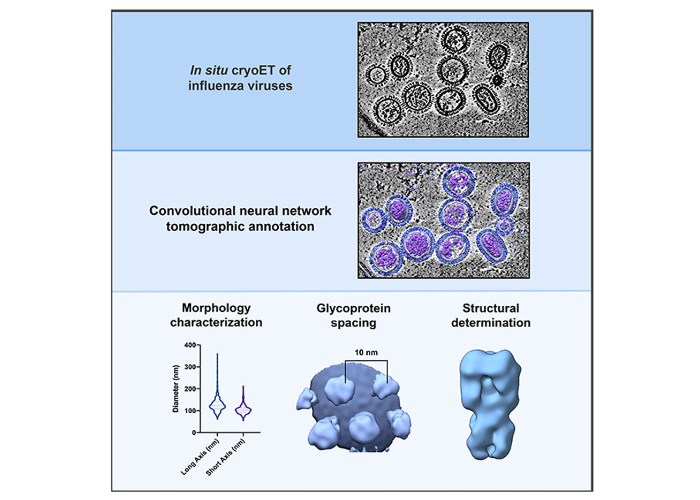
 Quantitative structural analysis of influenza virus by cryo-electron tomography and convolutional neural networks
Quantitative structural analysis of influenza virus by cryo-electron tomography and convolutional neural networksChancellor Collins to UMass Chan PhD candidates: ‘We need your science’
September 11, 2025



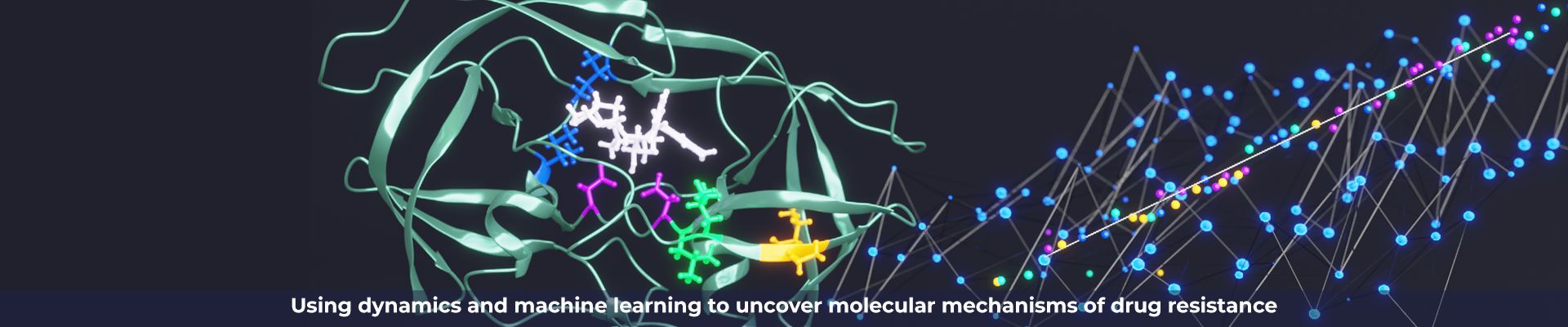
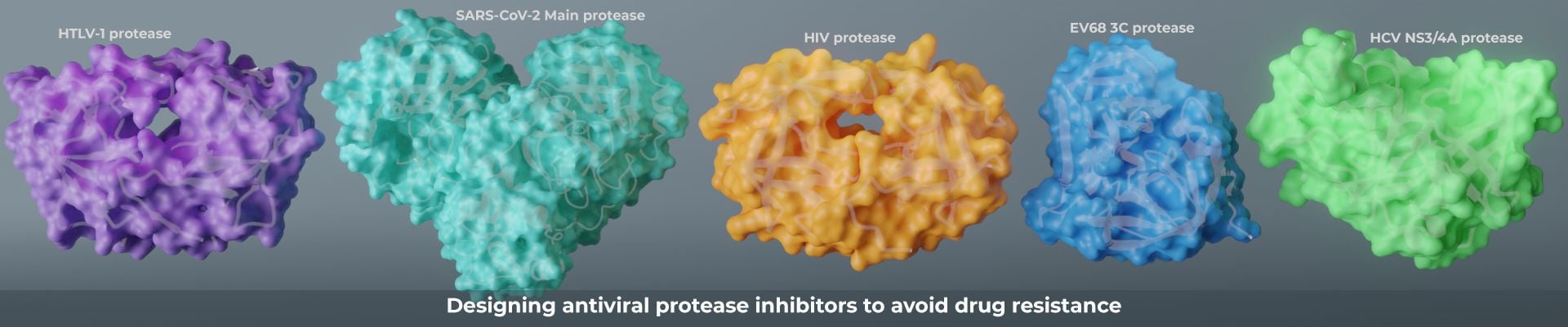
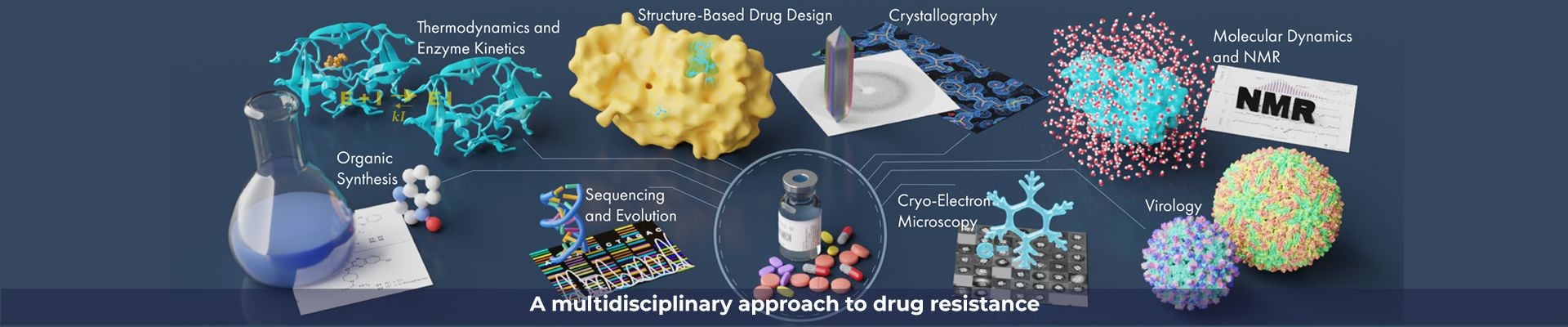
We combine a variety of experimental and computational techniques to understand the molecular basis of drug resistance and pay attention to the ways that the natural substrate specificity is maintained by the resistant viral variants. Our new paradigm of drug design minimizes chances of resistance.

We've realized that through understanding the molecular mechanism by which the disease process occurs we could develop inhibitors that block the disease in such a way that the likelihood of resistance occurring is greatly reduced.

We combine a variety of experimental and computational techniques to understand the molecular basis of drug resistance.

We are a collaborative team of scientists dedicated to understanding and thwarting drug resistance.
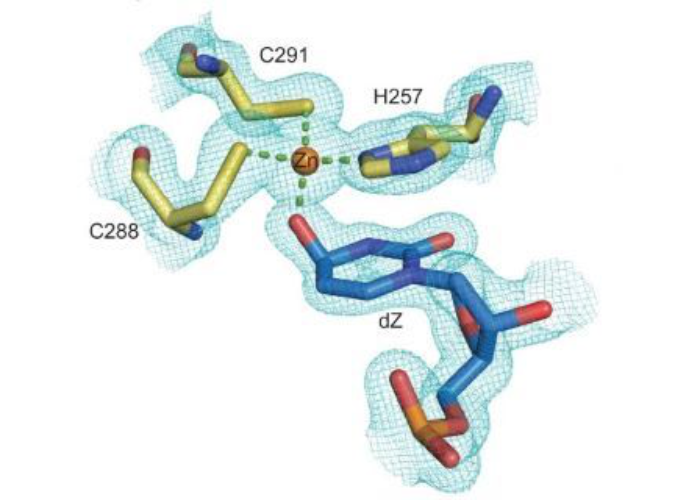
Maiti A, Hedger AK, Myint W, Balachandran V, Watts JK, Schiffer CA, Matsuo H. Nat Commun. 2022
oi: 10.1038/s41467-022-34752-1. PMID: 36402773; PMCID: PMC9675756.
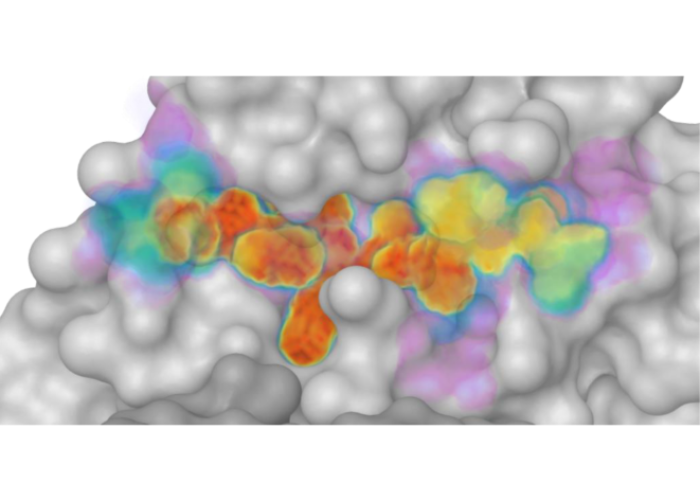
Defining the substrate envelope of SARS-CoV-2 main protease to predict and avoid drug resistance
Shaqra AM, Zvornicanin SN, Huang QYJ, Lockbaum GJ, Knapp M, Tandeske L, Bakan DT, Flynn J, Bolon DNA, Moquin S, Dovala D, Kurt Yilmaz N, Schiffer CA. Nat Commun. 2022
doi: 10.1038/s41467-022-31210-w. PMID: 35729165; PMCID: PMC9211792.
Qiuyu J. Huang, Kangkang Song, Chen Xu, Daniel N.A. Bolon, Jennifer P. Wang, Robert W. Finberg, Celia A. Schiffer, Mohan Somasundaran. Structure March 2022.
DOI: 10.1016/j.str.2022.02.014
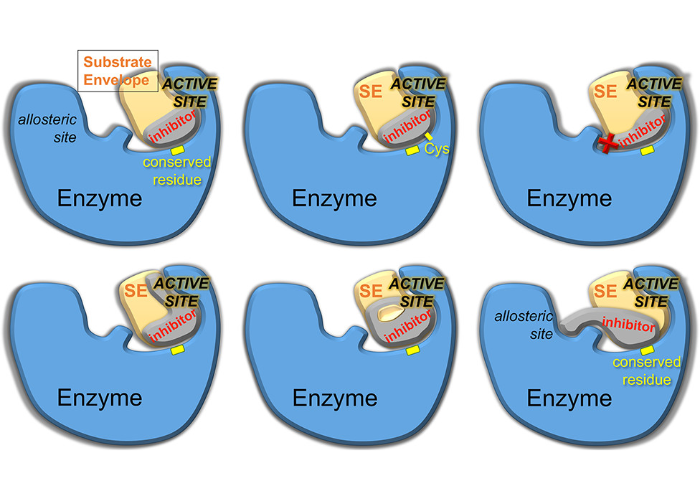 Drug Design Strategies to Avoid Resistance in Direct-Acting Antivirals and Beyond
Drug Design Strategies to Avoid Resistance in Direct-Acting Antivirals and Beyond











Molecular illustrations on this webpage were generated by Leonora Martínez-Núñez, PhD.