Lab Research


Retina
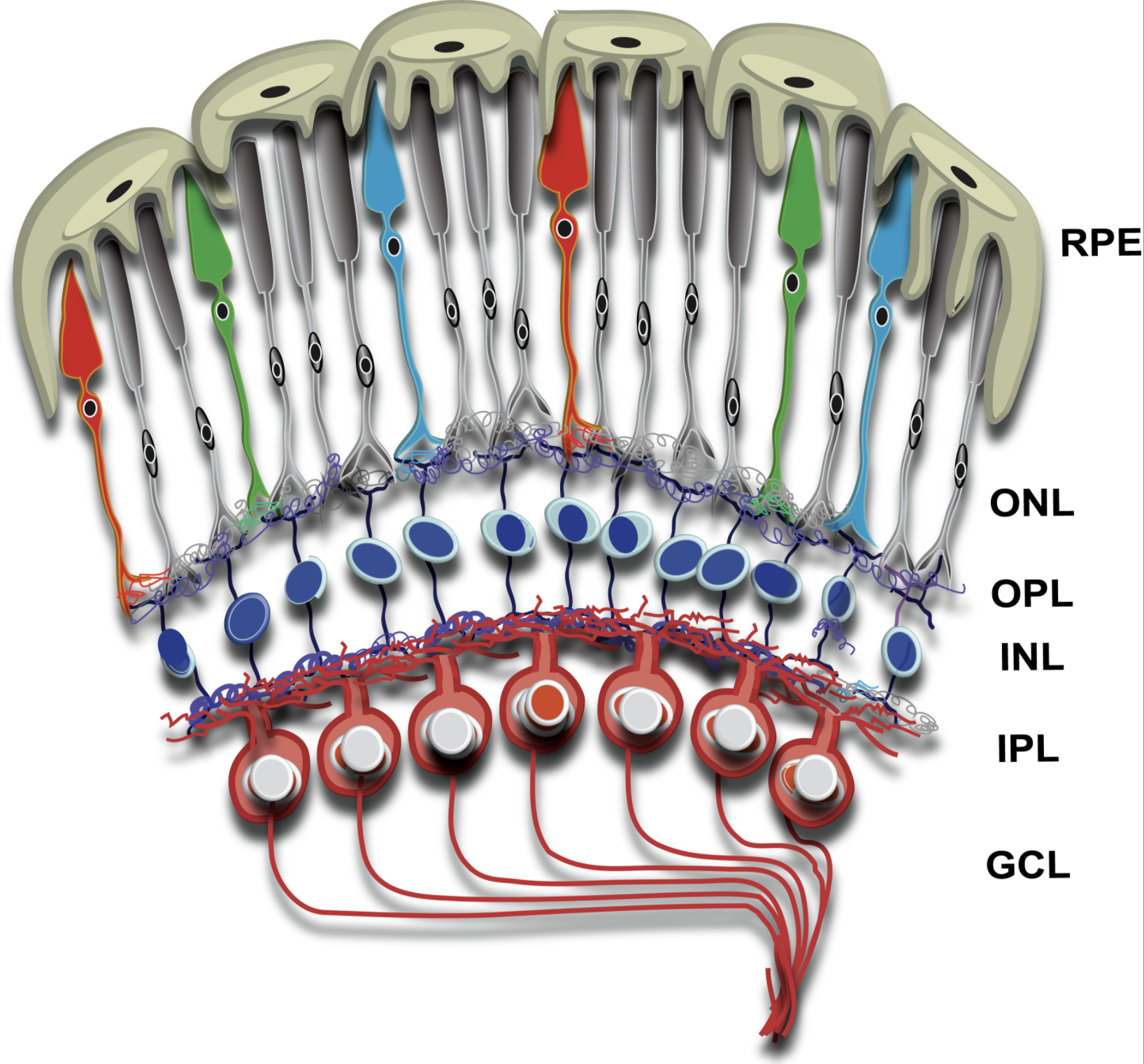
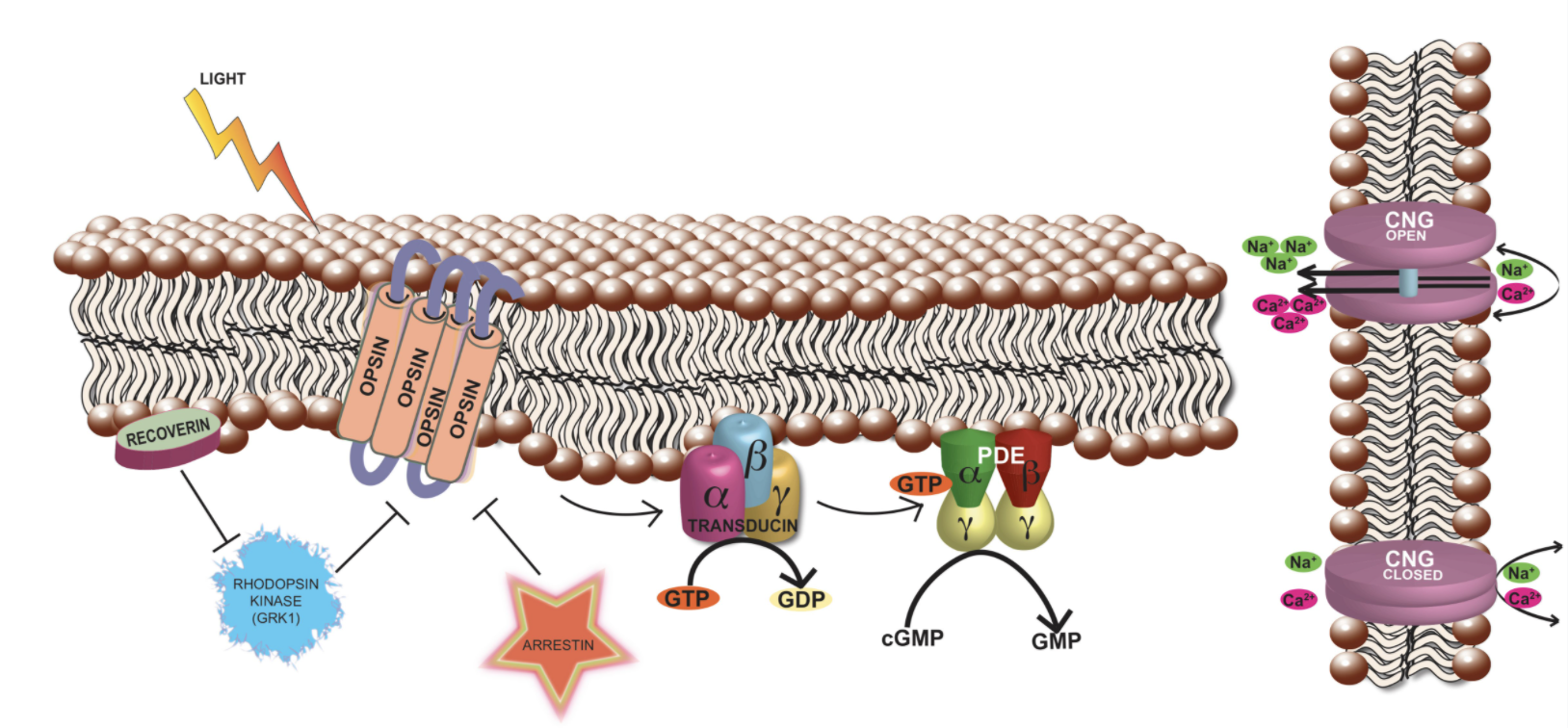
The retina is ~0.5 mm thick tissue situated in the back of the eye and is involved in the first steps of light sensation. It is a highly organized tissue consisting of six major types of neurons and one glial cell type separated by two synaptic layers, called the outer and the inner plexiform layers. Among the neurons, photoreceptors are the most abundant cell types and form the outermost layer of the retina. The tips of the photoreceptors are physically closest to the retinal pigment epithelium (RPE), which forms the outermost blood-retina barrier and is also involved in the visual cycle and periodic maintenance of the photoreceptor sensory compartment. The choroidal blood vessels overlying the RPE supply nutrients to photoreceptors.