Secondary Cone Death in Retinitis Pigmentosa
 One of the research focus in my group is on a family of inherited retinal degenerative diseases called Retinitis Pigmentosa, which cause massive loss of photoreceptors and consequently blindness. The pathology is characterized by an initial loss of night vision due to the loss of the night-active rod photoreceptors, followed by a progressive loss of the day-active cone photoreceptors. In many cases, the disease-causing mutation is in a gene that is exclusively expressed in the night-active rods; nonetheless, the day-active cones die too. To date, there is no known form of retinal degeneration in humans or mice where rods die, and cones survive. In contrast, mutations in cone-specific genes result only in cone death.
One of the research focus in my group is on a family of inherited retinal degenerative diseases called Retinitis Pigmentosa, which cause massive loss of photoreceptors and consequently blindness. The pathology is characterized by an initial loss of night vision due to the loss of the night-active rod photoreceptors, followed by a progressive loss of the day-active cone photoreceptors. In many cases, the disease-causing mutation is in a gene that is exclusively expressed in the night-active rods; nonetheless, the day-active cones die too. To date, there is no known form of retinal degeneration in humans or mice where rods die, and cones survive. In contrast, mutations in cone-specific genes result only in cone death.
Cones are essential for daylight, color, and high-acuity vision, thus it is their loss that leads to blindness. The fact that cone death always follows rod death independently of the underlying mutation in a rod-specific gene suggests that the reason for cone death might be similar across various forms of Retinitis Pigmentosa. Thus, targeting the common mechanism of cone death may allow for the development of vision therapies with broad clinical significance. This is particularly important because the number of genes that when mutated cause Retinitis Pigmentosa is quite large. Therefore, developing gene therapeutic approaches to treat mutations in rod-specific genes requires a logistically challenging number of clinical trials. Additionally, such therapies would only be effective if employed prior, or at the initial stages of the disease, meaning when almost all rods are still present and day light vision is nearly normal. In contrast, a therapy that intervenes at the level of cone death by either halting or delaying further degeneration can be applied at any stage of the disease progression and benefits all patients with Retinitis Pigmentosa. However, developing such a treatment using either gene therapeutic approaches or conventional drugs, requires a fundamental understanding of why cones die when the disease causing mutation is in a rod-specific gene.
We recently proposed that cones suffer from a nutrient shortage induced by the disruption of the retinal architecture once most rods have been lost (Punzo et al., 2009). Because rods outnumber cones at a ratio of >20:1 and photoreceptors account for approximately 75% of all retinal cells, loss of rods severely alters the retinal architecture. Thus we hypothesized that once rod death progresses beyond a critical threshold, cone death initiates as a cell autonomous event due to a reduction in nutrient flow from the adjacent cell layer, the retinal-pigmented epithelium. This hypothesis was based on the following findings: First, cone death always initiates only after approximately 90% of rods have died irrespective of the rod death kinetics. Second, during the period of cone degeneration cones display signs of prolonged starvation. Finally, during cone death, we observed gene expression changes in many metabolic genes and genes of the insulin/mammalian target of rapamycin (mTOR) pathway, a key pathway controlling cell metabolism. To test if activation of the insulin/mTOR pathway alters cone survival, we treated the fast progressing rd1 mouse model of Retinitis Pigmentosa with daily systemic injections of insulin. While cone survival did improve, the therapeutic effect of insulin lasted only for a period of four weeks possibly due to the negative feedback loop within the insulin/mTOR pathway.
 |
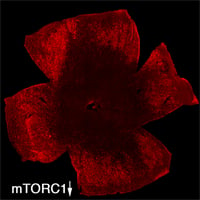 |
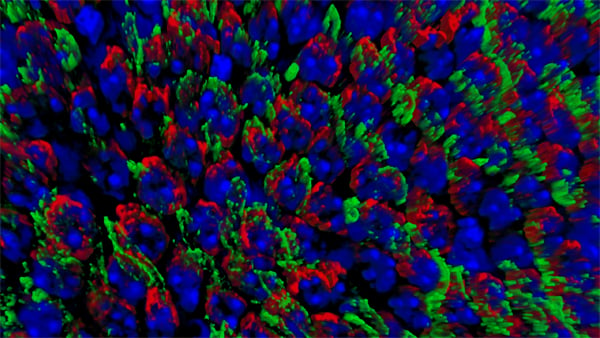
| Retinal Flat mount showing surviving cones in red at 2 months of age. Right image shows effect of loss of mTORC1 (fewer surviving cones at 2 months of age). Role over image to see the effect of increased mTORC1 activity on cone survival at 2 months of age. | |
To evaluate the long-term therapeutic potential of the insulin/mTOR pathway on cone survival, and test if insulin acted directly on cones through this pathway, we have now constitutively activated this pathway in cones through use of mouse genetics in two mouse models of Retinitis Pigmentosa. This has led to a robust and unprecedented cone survival effect in two mouse models of Retinitis Pigmentosa and to the finding that cone survival is driven solely by increased mTOR complex 1 activity (rollover image; Venkatesh et al., 2015). To study why the initial robust survival effect of constitutively activated mTORC1 does not persist indefinitely we further study the consequences of altered mTORC1 activity (Venkatesh et. al., 2016). Our studies show that it is not the strength of survival is directly dependent of the strength of mTORC1 activation but that the duration of the effect depends on the system's ability to intermittently turn of mTORC1. This knowledge will allow us to identify downstream genes of mTORC1 that mimic the positive effect of increased mTORC1 activity, without altering the systems ability to intermittently turn of mTORC1. In summary, the knowledge gained from these two studies will allow us to identify downstream target genes or pathways that when manipulated should extend vision in affected individual (see Jean Bennett commenting). For example, rAAV mediated gene transfer of mTORC1 targets in cones may provide a feasible approach for future treatments in humans. Alternatively, drugs that increase mTORC1 activity could be delivered to the retina through slow releasing eye implants. The focus of our current research is to identify the correct mTORC1 targets that can be used to prolong vision in humans with Retinitis Pigmentosa.
It is of interest to note that the most common form of photoreceptor degeneration and thus the major cause for blindness in the industrialized world, age-related macular degeneration (AMD), may result in photoreceptor death due to the same proposed reason by which cones die in RP. In AMD, retinal-pigmented epithelium cell function and health are affected as the accumulation of drusen between the retinal-pigmented epithelium and choriocapillary impinges on nutrient transfer from the bloodstream to retinal-pigmented epithelium cells. Because retinal-pigmented epithelium cells are the main source of nutrients for photoreceptors, a reduction in nutrient availability for retinal-pigmented epithelium cells inevitably affects photoreceptors too. Thus AMD and Retinitis Pigmentosa, although having different etiologies may both cause blindness by starving cones. Therefore, improving photoreceptor metabolism by increasing mTORC1 activity may be applicable not only to Retinitis Pigmentosa, but also holds great promise for treating the most common form of retinal degeneration in the industrialized world. Indeed our experiments suggest that by increasing mTORC1 activity it is possible to prolong photoreceptor survival once the retinal-pigmented epithelium has died (Zieger & Punzo 2016).
See Dr. Punzo and Aditya Venkatesh talk about
The role of mTORC1 in Retinitis Pigmentosa