Chromatin Structure and Function
Nucleosome positioning
The eukaryotic genome is packaged into a repeating subunit known as the nucleosome, which consists of 146 bp of DNA wrapped nearly twice around an octamer of basic histone proteins. All eukaryotic DNA transactions, from transcription to DNA repair to recombination, occur in the context of this nucleosomal packaging. As the DNA wrapped around the nucleosome and the DNA located between nucleosomes differ in their accessibility and structural characteristics, the precise locations of nucleosomes along the genome have proven to be of great importance for understanding the function of the genome. In some sense, one can think of the chromatin packaging of the genome as a “filter set” that affects genomic output by alternately hiding or exposing key regulatory DNA elements, allowing the same DNA molecule to express multiple distinct functional programs depending on its precise packaging state. Our laboratory has long been interested in the structural biology of the genome – what are the rules underlying genomic packaging, and what are the functional implications of different packaging states?
We have focused on the budding yeast S. cerevisiae as a convenient model organism to study chromatin structure genome-wide, where we have developed genome-wide methods for analysis of chromatin architecture. For instance, to generate genomic maps of nucleosome positioning, we make use of the fact that micrococcal nuclease (MNase) has preference for linker DNA over nucleosomal DNA – size separation of the DNA fragments remaining after MNase digestion results in a regular “ladder” with repeating unit size. Genome-wide analysis of the resulting mononucleosome-sized fragments by tiling array or by deep sequencing (MNase-seq) then provides a catalogue of nucleosome-protected regions (Yuan et al, Science 2005), revealing both locations of “well-positioned” nucleosomes that are present in the same location in most of the cell population, and locations of “fuzzy” regions where the nucleosome location varies between cells. Among the greatest surprises from these early nucleosome maps was the fact that the majority of nucleosomes (~70%) in budding yeast are relatively well-positioned, despite the long-understood fact that the vast majority of genomic DNA has little to no intrinsic thermodynamic preference for incorporation into nucleosomes. We have continued to investigate aspects of nucleosome positioning in budding yeast, using both traditional genetic approaches (Weiner et al, Genome Res 2010) as well as comparative genomic studies (Tsankov et al, PLoS Bio 2010, Hughes et al, Mol Cell 2012) to illuminate the forces responsible for nucleosome positioning in vivo.
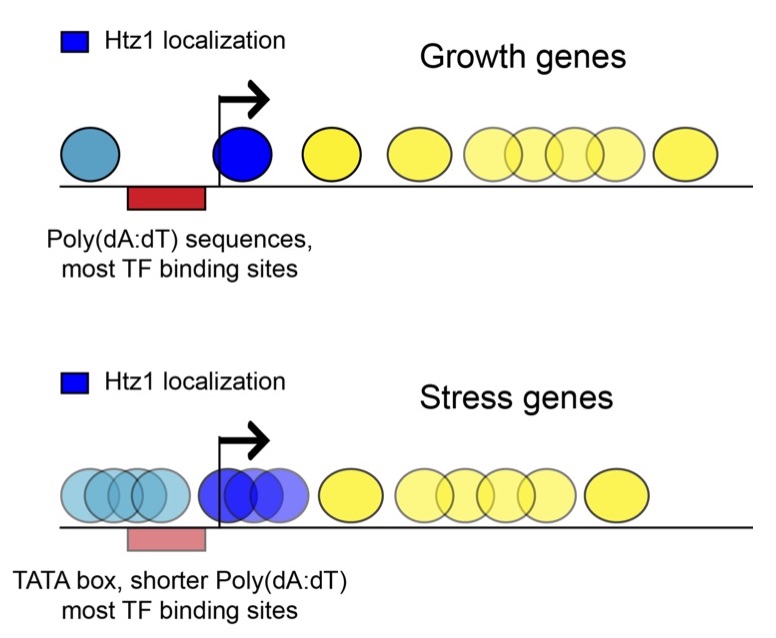
The lessons learned from yeast nucleosome maps have largely proved general in a multitude of species subject to genome-wide nucleosome mapping, with a number of key concepts emerging (Figure 1). First, active and poised promoters are nucleosome-depleted, as are active enhancers. Second, nucleosomes bordering regulatory elements are well-positioned. Nucleosomes are positioned with decaying precision at greater distances, leading to a fuzzier nucleosome template further away (~1000-2000bp) from boundary nucleosomes. Third, highly transcribed genes tend to have lower nucleosome occupancy and less precise positioning, most likely due to action of RNA polymerase II and its associated factors. Finally, the density of the nucleosome template (average distance between two adjacent nucleosomes) can vary, mostly due to trans factors. We continue to study nucleosome positioning to some extent in yeast, with an active interest in single molecule decomposition of ensemble measurements.
Histone dynamics
Epigenomic assays capture snapshots of complex cellular states. Unlike the DNA sequence, which is highly stable, chromatin can be highly dynamic, raising the question of the relevant time-scales that govern chromatin structures. This question is related to, but distinct from, the question of heterogeneity within a cell population. When we observe a “fuzzy” nucleosome in an ensemble measurement, does the location of the nucleosome change only at cell division (stable) or every few seconds (dynamic) – a wide range of time-scales is consistent with both the ensemble observation as well as the distribution of single-cell states. This question applies throughout the range of structures found in vivo, from long-range chromosomal interactions to nucleosome chemical modifications. Understanding the function of the structures observed will ultimately require knowing the relevant time-scales for the structures in question.
To characterize chromatin dynamics genome-wide, we have adapted genetically-encoded pule-chase methods to characterize replication-independent nucleosome turnover rates genome-wide (Dion et al, Science 2007). These studies reveal replication-independent turnover at transcribed genes, with particularly rapid histone replacement occurring at regulatory regions (promoters and enhancers). Mutant studies from many labs including ours have enabled mechanistic insights into the cellular machinery responsible for histone replacement.
Over longer time scales, the question of histone dynamics is key to any mechanistic understanding of how chromatin states are copied from one generation to the next. To understand the mechanism by which chromatin states could be inherited, it is necessary to understand the unique challenges posed by histone protein dynamics during replication. First, during genomic replication the passage of the replication fork disrupts histone-DNA contacts, and old histones must reassociate with daughter chromosomes at a location close to their original location on the mother chromosome. Otherwise, locus-specific epigenetic information would be randomly shuffled every generation. Second, old histones only account for one half of the histones on each new genome, and the remaining histones are newly synthesized during each S phase. This implies some information passage from old to new histones, as otherwise old chromatin states would rapidly be diluted by new histones.
To attempt to measure the extent of nucleosome movement during genomic replication, we made use of a genetic pulse-chase system developed by the van Leeuwen laboratory in which induction of Cre-Lox recombination results in a switch from one epitope-tagged histone H3 to a new epitope tag. This system enabled us to switch off an ancestral H3 tag and subsequently follow the genomic disposition of the ancestral H3 for several cell divisions (Radman-Livaja et al, PLoS Bio 2011). To our great surprise, we found that old histone proteins do not accumulate at epigenetically-regulated loci such as the subtelomeres, but instead accumulate at the 5’ ends of long, poorly-transcribed genes. As expected, old histones do not accumulate at loci exhibiting rapid histone turnover, but we also found that 3’ to 5’ movement of old histones along coding regions and histone movement during replication are required to explain the patterns of ancestral histone retention we observe. Most importantly, we find that old histones do not re-associate with daughter genomes at precisely the locus from which they dissociated, but instead that maternal histones stay within ~400 bp of their original location during replication, providing the first measure of this crucial parameter. Thus, any inheritance of chromatin states must occur at the scale of ~5-10 nucleosome domains rather than at single nucleosome resolution. These results therefore constrain the maximum amount of information theoretically carried by chromatin between generations.
Histone modifications
The beads on a string structure of the chromatin fiber is not composed of uniform beads, as the chemical makeup of individual nucleosomes can in principle be extremely variable from nucleosome to nucleosome, with important consequences for genome function. Most dramatically, the histone proteins can be chemically modified, with a prodigious variety of post-translational modifications occurring (acetylation, methylation, phosphorylation, ubiquitylation, and many more) at multiple residues in all of the histones. These chemical modifications can alter the chemical and physical properties of the nucleosome, but also serve to modulate binding by proteins that recognize the modified state (e.g., bind only to tri-methylated H3K4).
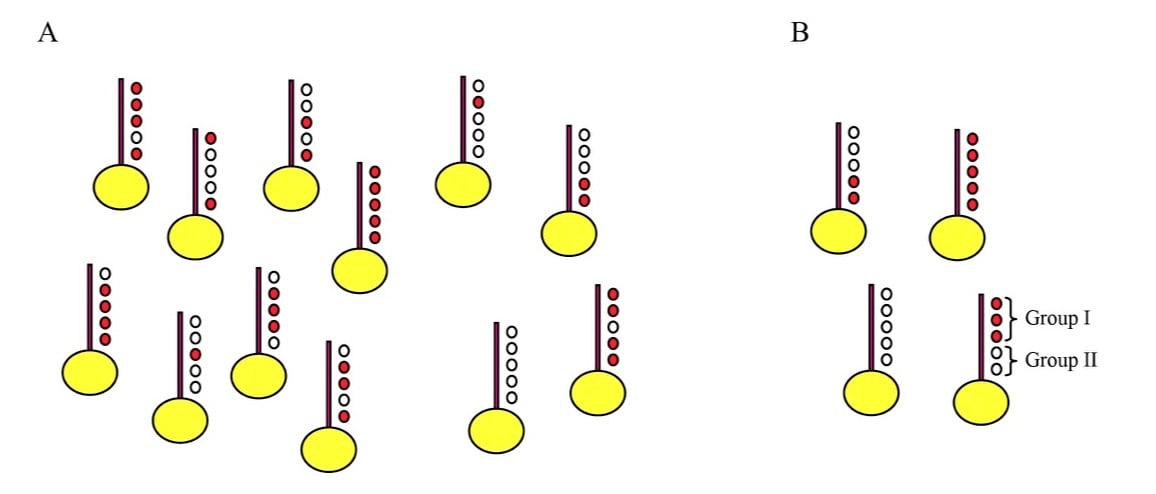
This incredible diversity of histone modifications leads naturally to the question of what it all means – why do so many histone modifications occur in the cell? We have long been interested in the question of how combinatorial complexity contributes to chromatin regulation. How many distinct modification combinations occur (Figure 2), and can specific combinations of modifications be matched with specific outcomes such that the code can be deciphered? We have taken two approaches to these questions, based on 1) genome-wide mapping of histone modifications at mononucleosome resolution (Liu et al, PLoS Bio 2005, Weiner et al, Mol Cell 2015), and 2) analysis of transcriptional changes in budding yeast carrying either mutant histones or deletions of histone-modifying enzymes (Dion et al, PNAS 2005, Weiner et al, PLoS Bio 2012).
Focusing first on genomic mapping, large-scale efforts in histone state mapping in multiple model systems reveal a number of conserved aspects of chromatin structure. First, the process of transcription leaves a massive footprint on chromatin, with different histone modifications deposited by the initiation (5’ end of genes) and elongation (middle and 3’ ends) forms of RNA polymerase. Marks at the 5’ ends of genes include H3K4 di/tri-methylation (H3K4me3), H3 and H4 tail hyper acetylation (H3K4/9/14/18/27ac and H4K5/8/12ac), H3.3 and H2A.Z variants, and H3K56 acetylation. Gene body nucleosomes are typically marked with H3K36me3 and H3K79me3, and are generally somewhat depleted of histone tail acetylations. Second, distal regulatory elements, such as enhancers, are marked with H3K4me1/2 but not H3K4me3. Other histone marks distinguish repressed, poised, and active enhancers, with H3K27 methylation and acetylation marking repressed and active enhancers, respectively. Third, two major forms of repressive chromatin are associated with specific histone modifications. Classical heterochromatin (including telomeres and many repetitive sequences) is marked with H3K9 methylation, while genes repressed by polycomb-group factors are marked with H3K27me3. H3K27-methylated nucleosomes can be either in a repressed state and lack other marks, or in a poised “bivalent” state in combination with the active mark H3K4me3. Finally, nucleosomes associated with centromeres often contain the H3-like CENP-A protein, and during M phase a broad pericentric domain of nucleosomes are marked with H3S10ph. Overall, there is a striking correspondence of histone state with the function of genomic regions, and because of this mapping of histone modifications is an effective method to discover genes and regulatory elements.
Turning to the functions of histone modifications, one systematic approach to understand the functions of combinatorial histone modifications has been whole genome analysis of gene expression changes observed in histone mutants. Such studies typically find that histone mutants exhibit relatively little phenotypic complexity resulting from different combinations of histone mutants. This is seen both in studies focusing on specific histone point mutants and their combinations, as well as in studies focusing on deletion mutants of histone modifying enzymes. For example, we systematically examined all 16 possible combinatorial mutations among the 4 lysines in the histone H4 tail, finding that mutation of three of the residues (lysines 5, 8, and 12) had indistinguishable effects on gene expression, whereas lysine 16 was confirmed to have unique effects on gene expression (Dion et al, PNAS 2005). Furthermore, gene expression defects in combinatorial mutants were little different from linear combinations of the component mutations – in other words, the effect of the H4K5,16R double mutant on gene expression could be predicted by adding the K5R and K16R datasets together.
In contrast to the modest effects of many histone modifying deletions observed at steady-state, single gene studies suggest that chromatin regulators have important roles in dynamic processes that are masked at steady-state. This has motivated us to study chromatin regulators in the context of transcriptional reprogramming, using diamide stress in yeast as a convenient paradigm for inducing massive transcriptional changes (Weiner et al, PLoS Bio 2012). We find that the majority of chromatin regulators have greater effects on gene induction/repression kinetics than they do on steady-state mRNA levels, confirming that dynamic studies can identify unanticipated functions for chromatin regulators. Grouping deletion mutants with similar gene expression defects identifies known complexes, and that joint analysis of histone mutants and deletion mutants associates many histone modifying enzymes with their target sites. By combining functional data with genome-wide mapping data, we identified a surprising role for Set1-dependent H3K4 methylation (an “activating” mark) in repression of ribosomal biogenesis genes. Together, these data provide a rich multi-modal view on the role of chromatin regulators in gene induction and repression dynamics, and suggest that understanding the myriad roles of chromatin structure in gene regulation on a genome-wide scale will require extending mutant analyses to kinetic studies.
Chromosome folding
While the 1-dimensional structure of chromatin is increasingly well-understood, our understanding of the three-dimensional folding of the genome in the nucleus has traditionally lagged behind. That said, over the past decade this gap has been dramatically narrowed thanks to the development by Dekker and colleagues of the 3C (Chromosome Conformation Capture) family of techniques. In these methods, chromatin is subject to crosslinking in vivo to capture interactions between chromosomal loci. The genome is then fragmented, typically with restriction enzymes, and DNA ligation is used to capture interactions between chromosomal loci that were in contact with one another in vivo. The abundance of ligation products between two chromosome regions is often interpreted as a measure of frequency/probability of contact, or proximity, between the pair of loci, providing a view of chromosome structure that is somewhat analogous to the view of protein structure generated by NMR techniques. Genome-wide variants of 3C, such as Hi-C, have revealed a number of organizational features of eukaryotic genomes at increasingly fine resolutions, from the scale of full chromosomal territories, to multi-Mb active and inactive compartments, to hundred-kb contact domains (TADs), to enhancer-promoter loops.
While many factors impact the effective resolution of a 3C/Hi-C dataset, including sequencing depth and library complexity, a crucial limit to genomic resolution is the size of the fragments generated before physical interactions are captured via ligation. Since the majority of 3C-based experiments rely on restriction enzymes for fragmentation of the genome, they are limited to ~1 kb resolution at best. To improve the resolution of 3C-based techniques, we therefore developed a high resolution 3C-based technique, dubbed “Micro-C”, in which fragmentation of the genome is accomplished using micrococcal nuclease (MNase) to enable mononucleosome-resolution analysis of chromosome folding. The improved resolution afforded by Micro-C enabled the identification of chromosomally-interacting domains – “CIDs” – in budding yeast that had not previously been appreciated using a restriction enzyme-based 3C technique (Hsieh et al, Cell 2015). A revised Micro-C protocol incorporating long crosslinkers, which we named “Micro-C XL”, not only recapitulated the local chromatin structures previously revealed by Micro-C, but also robustly recovered higher-order features such as centromere-centromere interactions (Hsieh et al, Nature Methods 2016).
Our method provides insight into yeast genome folding at all length scales of interest. At larger scales, the Rabl configuration of chromosomes is seen as clustering of centromeres, and interactions between the telomeres of chromosome arms of similar length. Centromeric chromatin also shows a characteristic “X” shape resulting from the two arms of the chromosome both statistically leading away from the centromere together in a hairpin-like structure for some ~20 kb, whereas centromeres are relatively isolated from chromosome arms. At higher resolution (Figure 3), genes in both budding and fission yeast are organized into chromosomally-interacting domains (CIDs), typically spanning 1-5 genes, that are in some ways similar to the “topologically-associating domains” described in a multitude of other model organisms. Boundaries between CIDs occur at active promoters, highly-expressed genes, and tRNA genes. The concordance of multiple independent approaches to analysis of chromosome folding provides hope that we are approaching a true understanding of the structures adopted by chromosomes in the cell.