


Free Energy Calculations
The drug-resistant protease variants have lower binding affinity to inhibitors than wild-type enzyme, while maintaining enough enzymatic activity for the virus to propagate. X-ray crystallography has been useful to elucidate the structural basis for the reduced affinity of drug-resistant variants for protease inhibitors. However, investigating binding energetics complements structural studies to arrive at a complete understanding of the critical components of differential binding affinity of resistant protease variants. We use free-energy decomposition to provide information about affinity changes due to specific kinds of interactions on an atomic level, which cannot be determined by experimental methods.
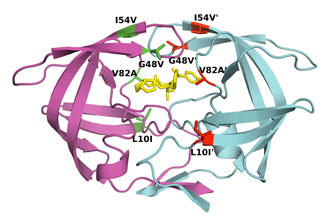
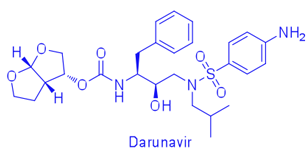
For example, difference in affinity for Darunavir (DRV) between wild-type (WT) protease and the V82T/I84V drug-resistant variant was calculated by free energy simulations and experimentally validated using isothermal titration calorimetry. The Gibbs free energy change for DRV binding is -15.2 kcal/mol for WT protease and -13.6 kcal/mol for V82T/I84V variant. Free energy computations revealed that the altered protease-inhibitor van der Waals interactions are the dominant component of the change in affinity due to V82T/I84V mutations. These comparative free energy computations for DRV binding to WT and drug-resistant proteases explain the energetic basis of resistance against this very high-affinity inhibitor, complementing structural findings. This combined methodology contributes to developing better strategies to design robust protease inhibitors. (Cai & Schiffer, 2010)