Projects
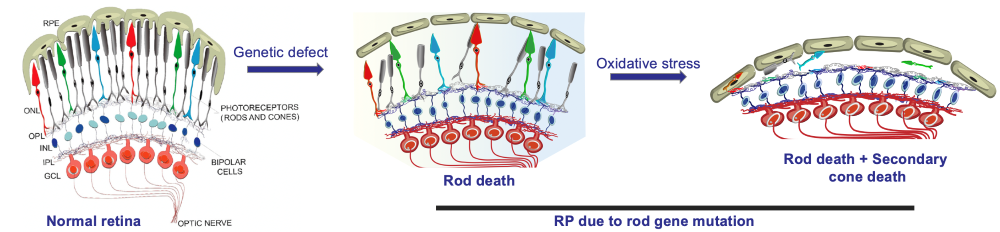
Project I: Retinitis Pigmentosa – X-linked RP – #1 Cause of RP
 Main Research Question: Understating RP mechanism in order to identify possible treatments for this disease.
Main Research Question: Understating RP mechanism in order to identify possible treatments for this disease.
Research Approach: Using known RP animal models (mice and zebrafish) to better understand the disease.
Concepts: "Growing retina in a petri dish"; using human skin cells from RP patients; convert to induced pluripotent stem cells (iPSCs) and then differentiate them into optic cups.
Collaborators: UPenn, UCL (University College of London)


Project II: Generating a Pig Model of Eyes Shut Homolog (EYS) Associated Retinitis Pigmentosa – #2 Cause of Autosomal Recessive RP
Main Research Question: There is no animal model for this genetic mutation. This project is focused on generating a pig model.

Research Approach: Using CRISPR-Cas9 to edit EYS in the genome and then examine the eye phenotype.
Concepts: Understand how the disease is caused; test new gene therapy in this model; conduct gene therapy safety studies in this model.
Collaborators: Dr. Heather Gray-Edwards in the Horae Gene Therapy Center and Dr. Jaime Rivera in the Department of Pediatrics UMass Chan
Project III: Minigene Therapy for Ocular Diseases Due to Mutations in Large Genes, Which Are Not Packageable in Conventional AAV
Main Research Question: How to deliver large genes for gene therapy using conventional AAV?
Research Approach: Generate shorter genes – "mini-genes" – which will be functional.

Concepts: To create a smaller (<4 Kbase), yet functional, form of the gene.
Collaborators: Dr. Guangping Gao in the Gene Therapy Center

Ongoing Minigene Therapy Programs:
- Usher Syndrome (RP), USH2A = Usherin → 15 Kbase
- Stargardt Disease (Degrative Retinal Disease) = ABCA4 → 8 Kbase
- EYS-Retinitis Pigmentosa (RP) → 9 Kbase

Project IV: Using the Knowledge From Rare Diseases to Approach Common Causes of Blindness
Main Research Question: Design a gene therapy to approach common blinding diseases (AMD, DR, ROP, neovascularization).
Research Approach: Reduce oxidative stress by delivering anti-oxidant genes;long-term anti-VEGF treatment.
Concepts: Use OXR-1 gene (anti-oxidative stress) to prolong  photoreceptors' life (mice); prolong cone survival in RP.
photoreceptors' life (mice); prolong cone survival in RP.
Collaborators: Dr. Michael Volkert in MAPS